
Chi ha detto che l’ADHD è roba da bambini? Per decenni, anche tra i professionisti della salute mentale, la risposta sarebbe stata quasi unanime: sì, è un disturbo dell’infanzia, qualcosa che si supera con l’adolescenza, come il morbillo o la paura del buio.
Eppure, i numeri raccontano un’altra storia. Circa il 2,5% degli adulti nel mondo convive oggi con un ADHD diagnosticabile, una percentuale che tradotta in persone reali significa milioni di individui che ogni mattina si svegliano cercando di gestire una mente che corre più veloce del mondo circostante, che perde il filo delle conversazioni, che dimentica appuntamenti importanti o che — all’opposto — si incolla a un’attività per ore perdendo la nozione del tempo.
Molti di loro non lo sanno nemmeno. Altri ci hanno messo anni, a volte decenni, per ottenere una diagnosi, spesso dopo aver collezionato frustrazioni lavorative, relazioni complicate e un senso pervasivo di “non stare al passo” che nessuna spiegazione riusciva a rendere conto.
Il distacco tra ciò che la scienza sa e ciò che la società percepisce è, in questo caso, abissale. L’ADHD ha attraversato un’intera odissea terminologica — da “danno cerebrale minimo” a “disfunzione cerebrale minima“, fino alle definizioni attuali di DSM-5-TR e ICD-11 — ma il concetto che si tratti di una condizione neurobiologicamente fondata, con basi genetiche e alterazioni cerebrali documentate, fatica ancora a sedimentarsi nell’immaginario collettivo.
Non è raro che adulti con ADHD brillanti e funzionali vengano liquidati con un “sei troppo intelligente per averlo” o, al contrario, che la loro difficoltà venga attribuita a pigrizia o mancanza di volontà. La validità scientifica dell’ADHD adulto, per fortuna, non è più in discussione tra chi studia il disturbo.
Ma la strada per far arrivare questa evidenza nella vita quotidiana delle persone è ancora lunga.

LA “VALIDITA'” dell’ADHD nell’ADULTO: 3 PILASTRI EMPIRICI
Quando un disturbo psichiatrico viene messo in discussione — e l’ADHD adulto lo è stato, con una virulenza che pochi altri disturbi hanno conosciuto — la scienza risponde con un concetto preciso: la validità. Non intendo la validità nel senso comune di “autenticità”, ma nel senso tecnico di “fondamento empirico”. Un disturbo è valido se possiede caratteristiche identificabili, se predice esiti specifici, se condivide basi biologiche con forme correlate. L’ADHD adulto soddisfa tutti e tre questi criteri, e lo fa con una robustezza che dovrebbe — ma non sempre riesce — a zittire i detrattori.
Validità descrittiva
Significa semplicemente che i sintomi dell’ADHD negli adulti sono riconoscibili, caratteristici e distinguibili dalla normale variabilità umana. Non si tratta di “essere distratti ogni tanto”, come tutti lo siamo.
L’adulto con ADHD presenta un pattern persistente — almeno sei mesi, in più contesti — di inattenzione che compromette il funzionamento:
- mente che vaga durante le riunioni,
- progetti lasciati a metà,
- difficoltà a seguire istruzioni complesse,
- disorganizzazione cronica che non migliora con la buona volontà.
L’iperattività, poi, si trasforma con l’età: il bambino che corre per la classe diventa l’adulto che non riesce a stare seduto tranquillo, che ha una sensazione di irrequietezza interiore, che parla eccessivamente o che si sente “girare” anche quando il corpo è fermo.
L’impulsività si manifesta in decisioni affrettate, in interruzioni conversazionali, in acquisti compulsivi o in cambi di lavoro improvvisi.
Questi sintomi non sono generici: studi transculturali hanno confermato una struttura bifattoriale — inattenzione e iperattività-impulsività — in campioni di adulti di diverse nazionalità, con specifici item che si mostrano in modo coerente attraverso le culture.
| Tipo di validità | Domanda chiave | Evidenza empirica per l’ADHD adulto |
|---|---|---|
| Descrittiva | I sintomi sono identificabili e caratteristici? | Pattern bifattoriale (inattenzione / iperattività-impulsività) confermato in adulti; sintomi differiscono qualitativamente dalla normale variabilità; pervasività in più contesti |
| Predittiva | Il disturbo predice esiti, corsi e risposte al trattamento? | Maggiore rischio di comorbilità psichiatriche e somatiche, disoccupazione, incidenti, comportamenti criminali; risposta specifica a stimolanti e atomoxetina |
| Concorrente | Esistono basi biologiche condivise con forme correlate? | Correlazione genetica >0.8 tra ADHD infantile e adulto; alterazioni neuroanatomiche simili; profili neuropsicologici sovrapponibili |
Validità predittiva
Un disturbo valido deve predire qualcosa di specifico: esiti a lungo termine, pattern di comorbilità, risposta a trattamenti. Gli adulti con ADHD hanno un rischio cinque volte superiore di sviluppare disturbi d’ansia, circa quattro volte e mezzo per la depressione maggiore, quasi nove volte per il disturbo bipolare, e circa cinque volte per i disturbi da uso di sostanze.
Ma non finisce qui: anche il corpo ne risente. L’obesità, l’asma, i disturbi del sonno, l’epilessia, le malattie cardiovascolari, persino il diabete di tipo 2 e — nei dati svedesi più recenti — l’Alzheimer e il Parkinson mostrano associazioni significative. Questo non è un profilo generico di “persona stressata”: è un pattern specifico di vulnerabilità che ha radici condivise, in parte genetiche, tra cervello e organo per organo.
E poi c’è la risposta ai trattamenti: gli stimolanti, che agiscono sui sistemi dopaminergici e noradrenergici, riducono i sintomi con effect size che vanno da 0,39 a 0,71 nei trial controllati — un’efficacia che non si osserva in individui senza ADHD trattati con la stessa molecola.
Validità concorrente
Un disturbo valido deve avere basi organiche condivise con le sue forme correlate o con condizioni simili. E qui l’ADHD adulto si rivela sorprendentemente coerente con la sua controparte infantile.
Gli studi di genetica molecolare mostrano una correlazione genetica superiore a 0,8 tra l’ADHD diagnosticata nei bambini e quella negli adulti: stessi loci a rischio, stessi pathway biologici, stessa impronta sullo sviluppo cerebrale precoce.
Le alterazioni neuroanatomiche — volumi ridotti in regioni cortico-sottocorticali, differenze nella materia bianca — si sovrappongono tra le due età, anche se tendono a attenuarsi con l’età in modo parallelo al miglioramento sintomatico.
I profili neuropsicologici mostrano deficit esecutivi simili: memoria di lavoro compromessa, variabilità del tempo di reazione, difficoltà di inibizione. Non stiamo parlando di due disturbi diversi che casualmente condividono un nome. Stiamo parlando di un’unica condizione neurobiologica che si esprime, con sfumature diverse, attraverso l’arco della vita.

GENETICA ED EREDITABILITÀ: QUANTO È “SCRITTO NEL DNA”?
L’ereditabilità dell’ADHD: i dati degli studi su gemelli e famiglie
Quando si parla di genetica e disturbi mentali, la prima domanda che sorge è quasi sempre la stessa: “Ma quanto è ereditario?”.
Per l’ADHD, la risposta è sorprendentemente alta. Tra i disturbi psichiatrici, l’ADHD si colloca nella fascia più alta per ereditabilità — un dato che, da solo, dovrebbe far desistere chi ancora lo considera un semplice “problema di educazione” o una scelta di stile di vita.
Gli studi su gemelli e famiglie, condotti per decenni su campioni pediatrici, convergono su una stima dell’ereditabilità compresa tra il 70% e l’80%. Questo significa che, in una popolazione data, la maggior parte della varianza nei sintomi ADHD è attribuibile a differenze genetiche tra gli individui, piuttosto che a fattori ambientali condivisi o non condivisi.
È un valore che mette l’ADHD sullo stesso piano della schizofrenia e del disturbo bipolare, e ben al di sopra di condizioni come la depressione maggiore o i disturbi d’ansia, dove l’ambiente gioca un ruolo proporzionalmente maggiore.
Ma ecco il punto cruciale: queste stime derivano prevalentemente da studi su bambini. E se l’ADHD adulto fosse — come qualcuno ha sostenuto — un fenomeno qualitativamente diverso, forse meno “genetico”? I dati dicono il contrario.
Quando gli studi su gemelli adulti utilizzano informanti multipli — non solo l’autovalutazione del paziente, ma anche le valutazioni di partner, familiari o amici — l’ereditabilità rimane superiore al 70%. Anzi, gli studi basati su diagnosi cliniche formali, piuttosto che su semplici questionari auto-compilati, riportano valori del tutto sovrapponibili a quelli pediatrici.
La differenza emerge invece quando si affidano esclusivamente all’autovalutazione: qui l’ereditabilità crolla al 30-40%, un artefatto metodologico che riflette la distorsione soggettiva del ricordo e della percezione, non una riduzione reale del carico genetico.
| Popolazione studiata | Metodo di valutazione | Ereditabilità stimata | Implicazione |
|---|---|---|---|
| Bambini e adolescenti | Genitori/insegnanti | 70-80% | Alto carico genetico, misurazione affidabile tramite osservatori esterni |
| Adulti (auto-riferimento) | Solo self-report | 30-40% | Sottostima artefatta; il ricordo personale introduce distorsione |
| Adulti (informanti multipli) | Self + partner/familiari | >70% | Il carico genetico è conservato; la validità della misurazione è cruciale |
| Adulti (diagnosi clinica) | Registri sanitari/interviste strutturate | >70% | L’ADHD clinicamente validata in età adulta mantiene la stessa ereditabilità |
C’è un’altra evidenza che collega l’ADHD infantile e quella adulta in un continuum genetico: la correlazione genetica di circa 0,5 tra le due forme. Questo valore, ottenuto da analisi cross-generazionali su ampie famiglie, significa che i fattori genetici che influenzano l’ADHD nel bambino si sovrappongono in buona parte — ma non completamente — con quelli che influenzano la persistenza o la riemersione dei sintomi nell’adulto. Non è la stessa identica cosa, ma c’è una continuità sostanziale.
Alcuni geni sembrano agire come “fattori di persistenza“, altri come “fattori di attenuazione” o di cambiamento fenotipico con l’età. L’iperattività motoria, ad esempio, tende a diminuire più marcatamente dell’inattenzione, e questa divergenza evolutiva ha probabilmente basi genetiche parzialmente distinte.
La natura dimensionale del ADHD
E poi c’è la natura dimensionale dell’ADHD, un aspetto che spesso sfugge nelle discussioni pubbliche. Noi tendiamo a pensare in categorie: o hai il disturbo, o non ce l’hai.
Ma la realtà genetica è più sfumata. La correlazione genetica tra ADHD clinicamente diagnosticato e sintomi ADHD nella popolazione generale — quella vasta fascia di persone che non ricevono mai una diagnosi ma che, se interrogate, riferiscono qualche difficoltà di concentrazione o impulsività — è stimata intorno a 0,6.
Questo significa che i geni che contribuiscono al disturbo vero e proprio sono in gran parte gli stessi che spiegano la distribuzione a curva gaussiana di questi tratti nella popolazione.
Non esiste una linea netta, nel DNA, tra “normale” e “patologico”. Esiste invece un gradiente, dove la diagnosi clinica rappresenta una soglia arbitraria — sebbene clinicamente utile — su un continuum biologico.
Questa dimensionalità ha implicazioni pratiche importanti. Significa che studiare l’ADHD clinicamente diagnosticato ci dice qualcosa di rilevante anche sui meccanismi della concentrazione e dell’impulsività nella popolazione generale. E viceversa: i tratti sub-clinici, quando studiati in grandi campioni, possono illuminare aspetti biologici che alimentano anche le forme più severe. È un dialogo bidirezionale tra clinica e ricerca di base che ha accelerato enormemente la comprensione del disturbo.
Ma l’ereditabilità, per quanto elevata, non è il fine della storia. È l’inizio. Dire che l’ADHD è “per il 70-80% genetico” non significa affermare un determinismo assoluto, né giustificare un atteggiamento rassegnato del tipo “tanto è scritto nel DNA”. Significa piuttosto che i fattori genetici creano una vulnerabilità, un terreno su cui l’ambiente agisce — potenziando, attenuando o, in alcuni casi, addirittura prevenendo l’espressione clinica del disturbo. E significa anche che, per capire davvero l’ADHD, dobbiamo scendere al livello molecolare: identificare quali varianti genetiche, comuni o rare, contribuiscono a questo elevato carico ereditario. È lì che la ricerca degli ultimi anni ha compiuto passi da gigante, e dove ci dirigiamo ora.
La genetica molecolare: GWAS e varianti rare
Se gli studi su gemelli ci dicono quanto la genetica conta, la genetica molecolare ci dice cosa conta — quali varianti specifiche, in quali geni, attraverso quali percorsi biologici. Qui la ricerca ha compiuto negli ultimi anni un balzo qualitativo, passando da ipotesi a scoperte sistematiche.
La meta-analisi GWAS più grande disponibile oggi — pubblicata nel 2023 su Nature Genetics — ha analizzato dati di 38.691 pazienti con ADHD e 186.843 controlli. Ma cos’è esattamente un GWAS?
L’acronimo sta per Genome-Wide Association Study, ovvero studio di associazione a livello genomico: una tecnica di indagine genetica che permette di esplorare sistematicamente l’intero genoma umano — tipicamente analizzando centinaia di migliaia o milioni di varianti genetiche chiamate SNP (single nucleotide polymorphism, polimorfismi a singolo nucleotide, cioè punti del DNA dove le persone differiscono per una singola “lettera” genetica) — per identificare quali di queste varianti siano statisticamente associate a un tratto o a una malattia.
In pratica, si confrontano i profili genetici di un grande gruppo di persone con una condizione (nel caso, l’ADHD) e un gruppo di controllo senza quella condizione, cercando varianti che compaiano più frequentemente nei primi rispetto ai secondi.
Il risultato? 27 loci genomici significativi, che implicano 76 geni candidati. Molti di questi geni sono upregolati — attivi a livelli più alti — durante le fasi precoci dello sviluppo cerebrale, in particolare nel periodo prenatale e perinatale. Non è un dettaglio marginale: conferma che l’ADHD ha radici nelle tappe fondamentali della costruzione del cervello, quando neuroni si differenziano, migrano e stabiliscono connessioni.
I percorsi biologici emergenti puntano verso sistemi dopaminergici e GABAergici, e verso un ruolo delle cellule gliali — quelle che un tempo consideravamo semplici “collanti” del tessuto nervoso, e che ora sappiamo essere attori attivi nella modulazione sinaptica.
| Livello di analisi | Scoperta chiave | Significato biologico |
|---|---|---|
| GWAS meta-analisi (38.691 casi / 186.843 controlli) | 27 loci significativi, 76 geni | Identificazione di varianti comuni a rischio; |
| Correlazione genetica bambini-adulti | >0,8 | Stesso background genetico di base; l’ADHD adulto persistente è geneticamente continuo con quello infantile |
| Variant rare (CNV, PTV) | Presenti nel 10-15% dei casi | Sovrapposizione con autismo e schizofrenia; effetti pleiotropici attraverso disturbi neuropsichiatrici |
| ADHD con esordio tardivo | Profilo poligenico diverso | Minore carico ADHD-specifico; maggiore correlazione genetica con depressione e abuso di sostanze |
E qui emerge un dato che chiarisce un dibattito durato anni: l’ADHD adulto è geneticamente la stessa cosa dell’ADHD infantile?
La risposta, per i casi persistenti, è sostanzialmente sì. La correlazione genetica tra ADHD clinicamente diagnosticata nei bambini e negli adulti supera 0,8 — un valore che indica sovrapposizione quasi totale del background genetico comune. Gli stessi loci a rischio, gli stessi pathway, le stesse impronte sul neurosviluppo. Questo non significa che non ci siano differenze fenotipiche con l’età — l’iperattività motoria si attenua, l’inattenzione persiste, la compensazione cognitiva entra in gioco — ma significa che, a livello genetico, stiamo guardando lo stesso disturbo che invecchia con la persona, non un’entità nosologica distinta.
Le varianti rare
Ma c’è un’altra faccia della genetica dell’ADHD, oltre alle varianti comuni identificate dai GWAS. Si tratta delle varianti rare — quelle che, per definizione, si trovano in una piccola percentuale della popolazione ma che possono avere effetti di gran lunga maggiori.
Le varianti protein-truncating (PTV), che interrompono la sintesi funzionale di una proteina, e le Copy Number Variants (CNV), delezioni o duplicazioni di segmenti cromosomici, sono presenti nel 10-15% degli individui con ADHD. È una percentuale non trascurabile, che diventa ancora più interessante quando si scopre che molte di queste CNV sono condivise con l’autismo e la schizofrenia — disturbi con esordio e presentazione diversi, ma con un sottostante substrato genetico di vulnerabilità neuropsichiatrica comune.
- Le PTV, o varianti che troncano la proteina, sono mutazioni nel DNA — tipicamente piccole delezioni, inserzioni o cambiamenti di singole lettere — che introducono un segnale di “stop prematuro” durante la lettura di un gene. Il risultato? La sintesi della proteina viene interrotta a metà, producendo un frammento incompleto, spesso instabile e privo della funzione originaria. È come se una ricetta di cucina venisse tagliata a metà pagina: il cuoco non sa più come completare il piatto. Queste varianti sono particolarmente rilevanti perché agiscono come “interruttori spezzati” in geni essenziali per lo sviluppo cerebrale.
- Le CNV, o varianti nel numero di copie, operano su una scala diversa: non alterano la sequenza di un gene, ma il suo “dosaggio”. Si tratta di delezioni (perdita di un segmento cromosomico, con riduzione della dose genica) o duplicazioni (guadagno di un segmento, con sovradosaggio) che possono coinvolgere intere regioni contenenti più geni. Immaginate un libro dove alcune pagine siano state fotocopiate due volte, o altre strappate via: il contenuto delle pagine rimanenti è intatto, ma la quantità di informazione è sbilanciata. Le CNV possono essere ereditate dai genitori o sorgere de novo (nuove) durante la formazione dei gameti o nelle prime divisioni embrionali.
Questa pleiotropia genetica — lo stesso allele che contribuisce a fenotipi diversi — spiega in parte perché, in famiglie con ADHD, non sia raro trovare casi di autismo, disturbo bipolare o psicosi, e viceversa.
Il cervello, dal punto di vista genetico, è un organo per il quale le distinzioni nosologiche nette faticano a trovare riscontro molecolare.
| Tipo di variante | Frequenza nell’ADHD | Caratteristiche | Sovrapposizioni |
|---|---|---|---|
| Variant comuni (GWAS) | Poligeniche, ciascuna di piccolo effetto | 27 loci, 76 geni; enrichment sviluppo cerebrale precoce | Depressione, disturbi d’ansia, tratti cognitivi |
| Variant rare PTV | ~10-15% dei casi | Effetti più grandi; interruzione funzionale di proteine | Autismo, schizofrenia (CNV condivise) |
| CNV (delezioni/duplicazioni) | ~10-15% dei casi | Segmenti cromosomici alterati; spesso de novo | Neurosviluppo atipico, ritardo intellettivo in alcuni casi |
ADHD ad esordio tardivo
E poi c’è il caso particolare — e controverso — dell’ADHD con esordio tardivo, quella che apparentemente emerge in adolescenza o addirittura in età adulta, senza storia clinica evidente nell’infanzia. I dati genetici qui raccontano una storia diversa.
Gli individui con esordio tardivo, o diagnosticati per la prima volta in età adulta, mostrano un profilo poligenico diverso: minore carico di varianti a rischio per ADHD specificamente, ma maggiore correlazione genetica con depressione e abuso di sostanze.
Sembra quasi che, in questi casi, l’ADHD emerga non tanto da una vulnerabilità del neurosviluppo precoce specifica, quanto dall’intersezione di altre vulnerabilità — affettive, comportamentali, legate all’autoregolazione — che si esprimono clinicamente solo quando le richieste ambientali superano le capacità compensatorie.
| Sottogruppo | Carico poligenico ADHD | Correlazione con depressione | Correlazione con abuso di sostanze | Interpretazione |
|---|---|---|---|---|
| ADHD infantile persistente | Alto | Moderata | Moderata | Vulnerabilità del Neurosviluppo stabile nel tempo |
| ADHD diagnosticata in età adulta (con sintomi sub-soglia infantili) | Moderato-alto | Moderata | Moderata | Continuità genetica con forma infantile, espressione ritardata |
| ADHD con esordio tardivo apparente | Basso-moderato | Alta | Alta | Fenocopia parziale; contributo maggiore di fattori affettivi e comportamentali |
Questo non significa che l’esordio tardivo “non esista” — traumi cerebrali, ad esempio, possono indurre sintomi ADHD de novo — ma suggerisce la necessità di una cautela diagnostica. Quando un adulto si presenta con sintomi ADHD senza alcuna traccia di difficoltà infantili, è probabile che oltre il 90% di questi casi, una volta applicate procedure differenziali rigorose, si riveli spiegabile da altre condizioni.
In sintesi, la genetica molecolare dell’ADHD disegna un quadro di notevole complessità ma anche di coerenza. Ma il DNA non è il destino: è il punto di partenza. Tra il genotipo e il fenotipo si intercalano epigenetica, ambiente, esperienze, scelte — un intreccio che la ricerca sta solo iniziando a disegnare.
Oltre il DNA: epigenetica, trascrittoma e microbioma
Il genoma è come una partitura musicale: contiene tutte le note, ma non dice nulla su come e quando verranno suonate. Per capire l’ADHD dobbiamo guardare oltre la sequenza del DNA, esplorando i meccanismi che regolano la sua espressione, i prodotti attivi che genera e persino gli ecosistemi microbici con cui interagisce. È un territorio affascinante, ma anche pieno di incertezze — dove le promesse superano spesso le evidenze consolidate.
L’epigenetica studia le modificazioni chimiche del DNA che alterano l’attività genica senza cambiare la sequenza nucleotidica. La metilazione del DNA è il meccanismo più studiato: gruppi metilici che si attaccano a specifici siti del genoma, silenziando o attivando geni come interruttori molecolari.
Per l’ADHD, sono stati condotti diversi studi EWAS (Epigenome-Wide Association Studies), che analizzano centinaia di migliaia di siti di metilazione in modo sistematico.
Mentre i GWAS esplorano le varianti della sequenza del DNA — le differenze di “lettere” genetiche tra individui — gli EWAS indagano le modificazioni epigenetiche, in particolare la metilazione del DNA. Questa è una marcatura chimica che avviene quando un gruppo metile (CH₃) si attacca a specifici siti del genoma, tipicamente a livello delle isole CpG (regioni ricche di coppie citosina-guanina).
La metilazione agisce come un interruttore regolatore: quando un gene è altamente metilato, la sua espressione viene generalmente silenziata o ridotta; quando è poco metilato, rimane attivo o upregolato. Crucialmente, la metilazione non altera la sequenza del DNA — le “lettere” restano le stesse — ma modifica come e quando un gene viene letto e tradotto in proteina. È un meccanismo attraverso cui l’ambiente lascia tracce sul genoma: fattori come stress prenatale, dieta, esposizioni tossiche, esperienze avverse precoci possono influenzare pattern metilazionali che persistono nel tempo e possono essere ereditati attraverso divisioni cellulari.
Il risultato, finora, è deludente: nessun sito di metilazione si è rivelato riproducibile attraverso più coorti indipendenti. Eppure, c’è un dettaglio significativo: il più grande studio EWAS condotto finora — con oltre 4.500 partecipanti — ha esaminato sintomi ADHD proprio in adulti, non in bambini.
Questo suggerisce che la ricerca epigenetica sta lentamente spostando il suo baricentro verso l’età adulta, consapevole che le modificazioni accumulate nel tempo potrebbero essere più informative di quelle misurate nei primi anni di vita. Ma serve cautela: l’epigenoma è tessuto-specifico e gli studi su ADHD utilizzano DNA isolato da sangue o cellule buccali — surrogati lontani dal tessuto cerebrale che realmente ci interessa. Interpretare questi dati è come cercare di capire un’orchestra ascoltando solo il violino.
| Approccio epigenetico | Campione più grande | Risultato chiave | Limite metodologico |
|---|---|---|---|
| EWAS ADHD (sintomi adulti) | N > 4.500 | Nessuna metilazione riproducibile tra coorti | DNA da sangue/cellule buccali, non tessuto cerebrale |
| EWAS ADHD (bambini) | Vari studi, N minori | Alcuni candidati interessanti, ma non replicati | Eterogeneità tra coorti, metodi di sequenziamento diversi |
Il Trascrittoma
Se l’epigenetica ci dice come i geni sono regolati, il trascrittoma ci dice cosa producono — l’insieme degli RNA messaggeri attivi in un dato momento e in un dato tessuto. Gli studi del trascrittoma nell’ADHD si dividono in due famiglie: quelli che confrontano il profilo di espressione genica nel sangue di persone con e senza ADHD, e quelli più sofisticati che integrano i dati GWAS con mappe di espressione tissutale — in particolare da diverse aree cerebrali e tipi cellulari — in studi TWAS (Transcriptome-Wide Association Studies).
I TWAS hanno identificato geni candidati nuovi, non catturati dai GWAS tradizionali, e hanno evidenziato percorsi biologici coerenti con la neurofarmacologia del disturbo: differenziazione dei neuroni dopaminergici, ciclo di rilascio della noradrenalina, attività lipasica sui trigliceridi — quest’ultima un’apertura intrigante verso il metabolismo lipidico, che potrebbe collegare l’ADHD ai rischi cardiovascolari e metabolici documentati negli studi epidemiologici.
Ma cos’è esattamente un TWAS? L’acronimo sta per Transcriptome-Wide Association Study, ovvero studio di associazione a livello del trascrittoma. È una tecnica di integrazione dati che colma il divario tra genetica e funzione biologica.
Il TWAS utilizza dati di espressione genica — misurati in tessuti specifici, come diverse aree cerebrali — per costruire modelli predittivi che stimano, per ogni individuo, quanto un dato gene sia espresso in quel tessuto, sulla base del suo profilo genetico. Poi confronta questi livelli predetti di espressione tra casi e controlli, identificando geni la cui espressione alterata è statisticamente associata al disturbo, anche se la variante genetica stessa non raggiungerebbe la significatività in un GWAS standard. In pratica, il TWAS chiede: dato che questa persona ha certe varianti genetiche, quanto dovrebbe esprimere questo gene nel suo cervello? E l’espressione predetta differisce da chi non ha ADHD?
Uno di questi studi TWAS ha anche confrontato direttamente l’ADHD diagnosticata nell’infanzia versus forme diagnosticate nell’età adulta, trovando geni differenzialmente associati — ma il campione era troppo piccolo per confermare statisticamente questa distinzione.
È un promemoria costante in questa ricerca: la complessità del disegno supera spesso la potenza statistica disponibile.
| Approccio trascrittomico | Tipo di campione | Scoperta chiave | Implicazione |
|---|---|---|---|
| Profili di espressione nel sangue | RNA da sangue periferico | Firme di espressione differenziali, ma non specifiche | Marker di stato, non necessariamente causali |
| TWAS integrati con GWAS | Dati di espressione da aree cerebrali | Pathway dopaminergico, noradrenergico, lipasico | Collegamento tra rischio genetico e funzione cerebrale; possibile ponte verso comorbilità metaboliche |
Microbioma Intestinale
E poi c’è il microbioma intestinale, il territorio più controverso e mediaticamente esploso degli ultimi anni. L’idea che i batteri del nostro intestino possano influenzare il cervello — l’asse intestino-cervello — ha conquistato l’immaginazione scientifica e pubblica. Per l’ADHD nell’adulto, una meta-analisi del 2024 ha aggregato dati da quattro studi caso-controllo, armonizzando metodi e analisi.
Il risultato? La diversità beta — la differenza nella composizione microbica tra individui con e senza ADHD — è associata alla diagnosi. Ma andando oltre, emergono associazioni specifiche: Ruminococcus torques risulta più abbondante nei soggetti ADHD e correlato con iperattività/impulsività; Eubacterium xylanophilum risulta meno abbondante. Entrambi i generi potrebbero modulare processi infiammatori, aprendo una finestra — ancora stretta — su possibili meccanismi patogenetici.
| Microbo | Associazione con ADHD | Meccanismo ipotizzato | Evidenza |
|---|---|---|---|
| Ruminococcus torques | ↑ Abbondanza | Legato a iperattività/impulsività; possibile modulazione infiammatoria | Meta-analisi di 4 studi, ma eterogeneità persistente |
| Eubacterium xylanophilum | ↓ Abbondanza | Ridotto in ADHD; possibile ruolo protettivo anti-infiammatorio | Stessa meta-analisi, risultato armonizzato ma non robusto |
| Diversità beta complessiva | Associata alla diagnosi | Composizione microbica globalmente alterata | Significativa, ma con forte eterogeneità tra coorti |
Tuttavia, un’ombra cala su questi dati: l’eterogeneità tra studi persiste anche dopo armonizzazione statistica. I metodi di sequenziamento differiscono, le popolazioni campionate variano per dieta, età, uso di farmaci, e i risultati non sempre convergono.
È un campo in cui l’entusiasmo deve essere temperato dalla consapevolezza che, al momento, non possiamo affermare con sicurezza che alterazioni del microbioma causino l’ADHD, né che modificarlo — con probiotici, prebiotici o diete — possa curarlo.
L’associazione c’è, ma la direzione causale (l’ADHD altera il microbioma attraverso stili di vita, o viceversa?) e la replicabilità restano domande aperte.
| Livello di analisi “omica” | Stato dell’arte per ADHD adulto | Promessa | Limite critico |
|---|---|---|---|
| Epigenetica (EWAS) | Nessun risultato riproducibile; campo emergente | Tracce di esperienza e ambiente nel DNA | Tessuti non cerebrali; potenza statistica insufficiente |
| Trascrittoma (TWAS) | Geni e pathway nuovi identificati; differenze bambino/adulto suggerite | Ponte tra genetica e funzione cerebrale | Campioni limitati per sottogruppi; replicazione necessaria |
| Microbioma | Associazioni specifiche ma eterogenee | Possibile bersaglio terapeutico non farmacologico | Causalità non stabilita; confondenti ambientali massicci |
Cosa emerge da questo panorama?
Che la biologia dell’ADHD nell’adulto non si esaurisce nel DNA, ma si estende a strati regolatori, prodotti genici e interazioni ospite-microbioma che costituiscono un ecosistema di vulnerabilità molto più complesso della semplice sequenza genetica. È un campo giovane, dove ogni scoperta deve essere pesata contro la mancanza di replicabilità e la distanza tra tessuto studiato e tessuto rilevante. Ma è anche un campo che promette, nel medio termine, di collegare i punti tra genetica, neurobiologia e possibili interventi — farmacologici o meno — personalizzati sul profilo biologico individuale.
NEUROSCIENZE E DOPAMINA: IL CERVELLO SOTTO L’ADHD
Neuropsicologia: l’ipotesi della disfunzione esecutiva
C’è un’osservazione clinica antica quanto affascinante: le persone con lesioni acquisite ai lobi frontali — da traumi, tumori, ictus — sviluppano una “sindrome disesecutiva” che assomiglia in modo impressionante all’ADHD caratterizzata da :
- difficoltà a pianificare,
- a inibire risposte inadeguate,
- a usare la memoria di lavoro per guidare il comportamento in modo finalizzato,
- a mantenere prestazioni consistenti nel tempo.
Questo parallelo, notato già negli anni ’80, ha spinto decenni di ricerca volta a isolare un “profilo” neuropsicologico dell’ADHD — una firma cognitiva che, si sperava, potesse illuminare i substrati neurali, guidare interventi riabilitativi e persino aiutare la diagnosi.
Le meta-analisi condotte su adulti con ADHD confermano deficit in diversi domini esecutivi, con una coerenza che non può essere attribuita al caso:
- La decision-making risulta compromessa — non nel senso di incapacità di scegliere, ma di tendenza a preferire ricompense immediate a benefici futuri, di difficoltà a valutare rischi in modo ponderato.
- La memoria di lavoro, quel sistema che ci permette di tenere attiva e manipolare informazione per compiti in corso, mostra deficit robusti e riproducibili.
- L’attenzione sostenuta e focalizzata è altrettanto vulnerabile, così come la fluenza verbale — la capacità di generare rapidamente parole appartenenti a una categoria — e lo shifting, ovvero la flessibilità nel passare da un compito mentale a un altro.
- La memoria verbale a lungo termine, infine, emerge anch’essa compromessa in alcune analisi, suggerendo che il deficit non si limita ai processi “online” ma si estende alla consolidazione e al richiamo.
| Dominio cognitivo | Deficit documentato in meta-analisi adulte | Analogia con sindrome disesecutiva frontale | Effetto del trattamento |
|---|---|---|---|
| Memoria di lavoro | ↓ Capacità di mantenere/manipolare informazione attiva | Classico deficit da lesioni frontolaterali | Metilfenidato ↑; atomoxetina = |
| Attenzione sostenuta/focalizzata | ↓ Vigilanza nel tempo, distraibilità | Instabilità della performance da danno frontale | Metilfenidato ↑; atomoxetina ↑ |
| Decision-making | ↓ Preferenza ricompensa immediata, ↓ valutazione rischi | Disinibizione e impulsività da lesioni orbitofrontali | Metilfenidato ↑; atomoxetina ↑ |
| Inibizione della risposta | ↓ Capacità di sopprimere risposte prepotenti | Deficit classico da danno frontale | Metilfenidato ↑; atomoxetina ↑ |
| Fluenza verbale | ↓ Generazione rapida di parole | ↓ Flessibilità cognitiva associata a frontale | Metilfenidato ↑; atomoxetina ↑ |
| Shifting (flessibilità) | ↓ Passaggio tra set cognitivi | ↓ Flessibilità da lesioni frontali | Metilfenidato ↑; atomoxetina ↑ |
| Memoria verbale a lungo termine | ↓ Consolidazione/richiamo | Deficit associati a circuiti fronto-limbici | Metilfenidato ↑; atomoxetina ↑ |
Ma ecco il punto che ogni clinico — e ogni paziente — dovrebbe tenere a mente: nessun test neuropsicologico, da solo o in batteria, diagnostica l’ADHD.
Questo non è un difetto della ricerca, ma una caratteristica del disturbo. I test esecutivi possono distinguere, a livello di gruppo, persone con ADHD da controlli sani, ma non riescono a distinguere l’ADHD da altre condizioni cliniche — disturbo bipolare, depressione, disturbi d’ansia, persino alcune forme di demenza iniziale — che possono presentare profili sovrapponibili.
La NICE britannica, dopo revisione sistematica della letteratura e consultazione con esperti e pazienti, ha esplicitamente sconsigliato l’uso di test neuropsicologici come strumento diagnostico stand-alone nell’adulto, ovvero non associato ad una valutazione clinica più ampia e integrata.
Eppure, dire che i test neuropsicologici non servono alla diagnosi non significa dire che non servano per niente. Al contrario: il loro valore risiede nel profilare il pattern cognitivo individuale, identificando punti di forza da sfruttare e aree di vulnerabilità da supportare.
- Un adulto con ADHD può avere una memoria di lavoro discreta ma un’attenzione sostenuta gravemente compromessa, o viceversa.
Conoscere questo profilo aiuta a orientare scelte educative, professionali, terapeutiche. Aiuta a capire perché certi ambienti lavorativi — quelli strutturati, con feedback immediati, con varietà di compiti — funzionano meglio di altri. E aiuta a spiegare al paziente che le sue difficoltà non sono “pigrizia” o “mancanza di volontà”, ma riflettono pattern neurali misurabili e, in parte, modificabili.
| Uso dei test neuropsicologici | Appropriato | Non appropriato |
|---|---|---|
| Profilazione cognitiva individuale | ✓ Guidare trattamento, educazione, scelte occupazionali | ✗ Come sostituto dell’intervista clinica strutturata |
| Monitoraggio dei cambiamenti nel tempo | ✓ Valutare risposta a trattamenti o interventi | ✗ Come screening di popolazione |
| Psicoeducazione del paziente | ✓ Normalizzare, spiegare pattern di difficoltà | ✗ Come “prova” oggettiva per diagnosi differenziale |
| Ricerca e comprensione dei meccanismi | ✓ Identificare sottogruppi neuropsicologici | ✗ Per escludere/confirmare diagnosi da soli |
Un aspetto particolarmente illuminante riguarda l’effetto dei farmaci su queste funzioni cognitive.
Una meta-analisi recente ha esaminato gli effetti della somministrazione — almeno tre giorni — di stimolanti (metilfenidato) e non-stimolanti (atomoxetina) sui domini esecutivi.
- Il metilfenidato migliora significativamente tutti i domini testati, con l’effetto più marcato sull’attenzione e il più modesto sul tempo di reazione globale.
- L’atomoxetina, a sua volta, mostra benefici su tutti i domini tranne uno: la memoria di lavoro, dove l’effetto non raggiunge la significatività statistica.
Questa discrepanza farmacologica è intrigante: suggerisce che la noradrenalina da sola — il bersaglio principale dell’atomoxetina — non è sufficiente per ottimizzare tutti i circuiti esecutivi, e che il sistema dopaminergico, potenziato dai stimolanti, svolge un ruolo specifico nella memoria di lavoro prefrontale. Un dato, questo, che non solo affina la comprensione dei meccanismi, ma potrebbe orientare scelte terapeutiche personalizzate in futuro.
Neuroimaging: cosa cambia nel cervello ADHD?
Se i test neuropsicologici ci raccontano cosa funziona diversamente, l’imaging cerebrale ci mostra dove — o almeno cerca di farlo. La ricerca neuroanatomica sull’ADHD ha attraversato fasi entusiastiche e deludenti, promesse e ritrattazioni, ma sta lentamente convergendo verso un quadro più maturo e, soprattutto, più consapevole dei propri limiti.
La storia della materia grigia è forse la più nota.
La Materia Grigia
Meta-analisi strutturali su campioni pediatrici hanno riportato, per anni, alterazioni diffuse: volumi ridotti in regioni cortico-sottocorticali coinvolte nell’attenzione, nel controllo motorio, nella regolazione emotiva — corteccia prefrontale, gangli della base, talamo, cervelletto. Il dato interessante, però, è quello che accade con l’età.
Queste differenze, evidenti nei bambini, tendono a ridursi o scomparire negli adulti, in un parallelo sorprendente con il miglioramento sintomatico che molti individui sperimentano naturalmente. N
Non è una coincidenza: suggerisce che almeno parte delle alterazioni strutturali rappresenti un ritardo maturativo, piuttosto che un danno fisso. Il cervello ADHD, in altre parole, non è necessariamente “diverso” in modo permanente: è “più lento” a raggiungere la maturazione, e quando lo fa — spesso verso la tarda adolescenza o i primi vent’anni — i sintomi tendono a attenuarsi.
Ma la materia grigia non è tutto il cervello. La materia bianca, i fasci di assoni mielinici che connettono regioni distanti, racconta una storia parzialmente diversa.
La Materia Bianca
Qui le meta-analisi di studi con diffusion-weighted imaging — una tecnica che misura la direzionalità del movimento dell’acqua, surrogato dell’integrità dei fasci — hanno evidenziato differenze nel corpo calloso, la grande struttura che connette i due emisferi. E, a differenza della materia grigia, queste differenze appaiono più evidenti negli studi che includono adulti che in quelli limitati ai bambini.
È un contrasto affascinante, che ha spinto gli studiosi a ipotizzare traiettorie di sviluppo distinte: la materia grigia mostrerebbe un ritardo maturativo più marcato in età pediatrica, mentre la materia bianca esprimerebbe la sua atipia più tardi, forse perché la mielinizzazione — processo che continua fino alla terza decade di vita — segue tempi diversi.
Sono ipotesi, per ora. Gli studi longitudinali che potrebbero confermarle, seguendo gli stessi individui nel tempo, sono pochissimi.
| Tipo di tessuto | Pattern nei bambini | Pattern negli adulti | Interpretazione ipotetica |
|---|---|---|---|
| Materia grigia | Alterazioni diffuse, volumi ridotti cortico-sottocorticali | Differenze ridotte o assenti | Ritardo maturativo che si risolve parzialmente con l’età |
| Materia bianca | Differenze meno consistenti | Alterazioni del corpo calloso più evidenti | Traiettoria di mielinizzazione atipica, espressione più tardiva |
Di fronte a questi dati, però, bisogna ammettere una verità scomoda: la letteratura neuroanatomica sull’ADHD è sorprendentemente eterogenea e la convergenza è limitata. Questo non è necessariamente perché “non c’è nulla da vedere”, ma perché vedere richiede metodi comparabili — e qui la ricerca ha peccato di frammentazione.
Protocolli di acquisizione MRI diversi, percorsi e approcci non standardizzati, criteri di inclusione variabili (con o senza comorbilità, con o senza trattamento farmacologico, diverse età, diversi IQ), analisi statistiche disomogenee. Tutto questo “rumore metodologico” sovrasta in parte il “segnale biologico”.
C’è anche un’eterogeneità clinica, oltre a quella metodologica, che complica ulteriormente il quadro.
L’ADHD non è un’entità uniforme: le presentazioni cliniche differiscono (inattenzione pura versus combinata), il sesso modula l’espressione fenotipica e forse anche quella neuroanatomica, e il trattamento con stimolanti può alterare — in modo reversibile — i volumi cerebrali.
Alcuni studi suggeriscono che le alterazioni strutturali siano più pronunciate nei casi con comorbilità o con sintomi persistenti, mentre altri non replicano questa associazione. La maggior parte della ricerca, inoltre, si è limitata al confronto casi-controlli, perdendo l’opportunità di collegare le caratteristiche cerebrali individuali a esiti clinicamente rilevanti — come la persistenza dei sintomi nel tempo, o la risposta al trattamento. È un po’ come avere fotografie di gruppo e volere ritratti individuali: la tecnologia c’è, ma il campione e il disegno degli studi spesso non consentono quel livello di granularità.
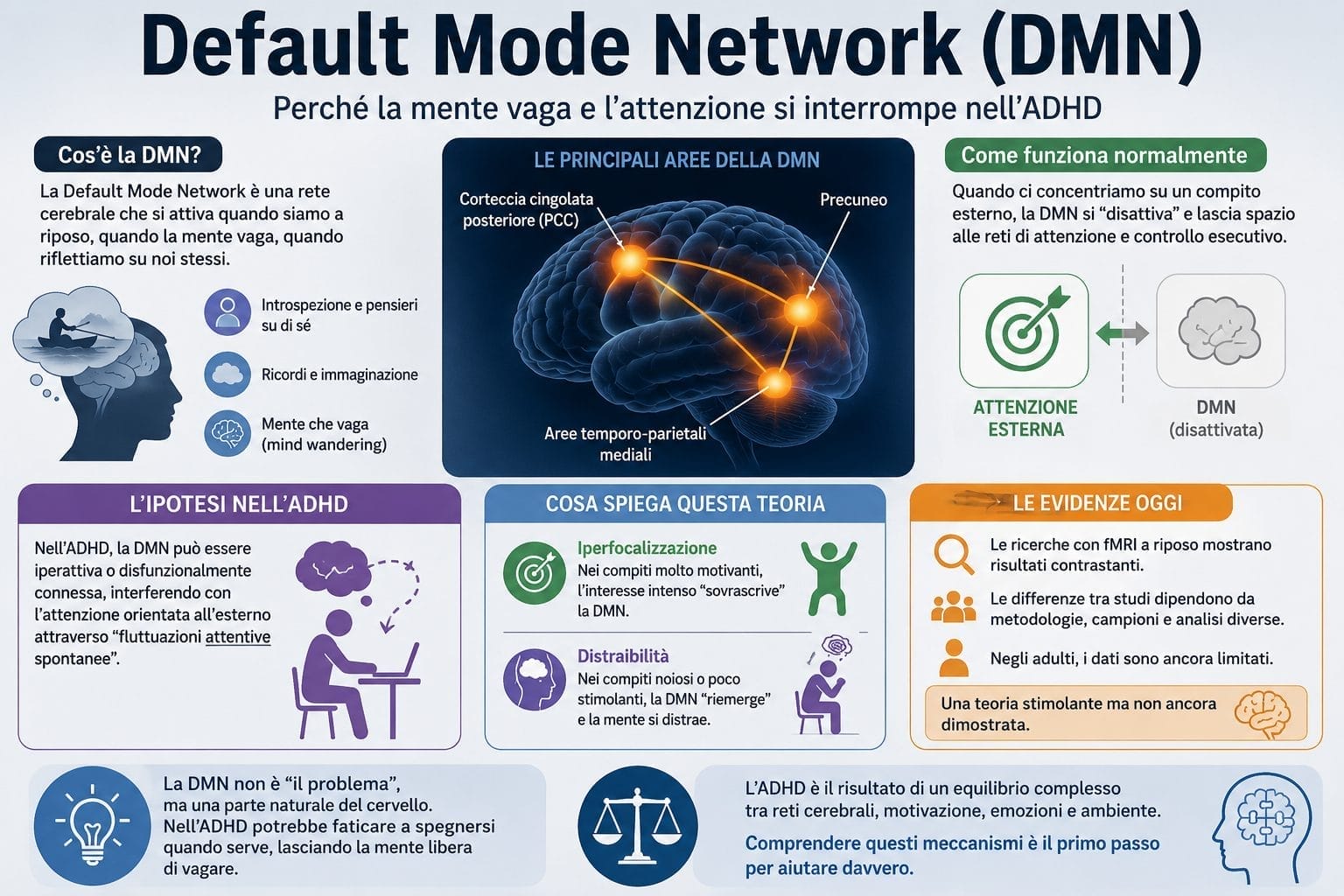
Default Mode Network (DMN)
Dal punto di vista funzionale, uno dei modelli più intriganti — e controverso — è la teoria della default mode network (DMN).
Questa rete cerebrale, che include regioni come la corteccia cingolata posteriore, il precuneo e le aree temporoparietali, è normalmente attiva quando siamo a riposo, quando la mente vagabonda, quando riflettiamo su noi stessi.
Quando ci concentriamo su un compito esterno, la DMN dovrebbe “disattivarsi”, cedendo il passo a reti di attenzione e controllo esecutivo. L’ipotesi, formulata da Sonuga-Barke e Castellanos, è che nell’ADHD la DMN sia iperattiva o disfunzionalmente connessa, interferendo con l’attenzione orientata all’esterno attraverso “fluttuazioni attentive spontanee” — quei momenti in cui, durante una lettura o una conversazione, la mente si stacca bruscamente dal presente e divaga verso pensieri irrilevanti.
È un’idea elegante, che spiegherebbe fenomeni come l‘iperfocalizzazione in compiti altamente motivanti (quando la DMN viene “sovrascritta” dall’interesse intenso) e la difficoltà a mantenere l’attenzione in compiti noiosi (quando la DMN “riemerge” incontrastata).
Ma le evidenze empiriche sono, al momento, contrastanti. Due meta-analisi di studi di fMRI a riposo in bambini, adolescenti e adulti hanno raggiunto conclusioni divergenti, probabilmente a causa di differenze metodologiche nelle analisi di connettività e nella definizione stessa dei nodi della rete. Per gli adulti, in particolare, i dati sono ancora troppo scarsi per trarre conclusioni definitive. La teoria della DMN resta, quindi, un’ipotesi stimolante ma non dimostrata — un promemoria che nella neuroscienza, come in ogni scienza, l’eleganza concettuale non garantisce la verità empirica.
| Teoria | Meccanismo proposto | Evidenza per ADHD adulto | Stato |
|---|---|---|---|
| Default Mode Network iperattiva | DMN non si disattiva durante compiti esterni, causando distrazione interna | Meta-analisi con risultati contrastanti; dati adulti insufficienti | Ipotetica, da verificare |
| Ritardo maturativo (materia grigia) | Sviluppo corticale più lento, specialmente frontale | Supportato da studi longitudinali pediatrici, meno dati adulti | Parzialmente confermato |
| Atipia maturativa (materia bianca) | Mielinizzazione anomala, specialmente interemisferica | Evidenze più forti negli adulti che nei bambini | Emergente, richiede studi longitudinali |
| Eterogeneità neurobiologica | Sottogruppi con profili neurali distinti | Supportato da analisi di cluster, ma non ancora clinicamente utilizzabile | Promettente per medicina di precisione |
Cosa possiamo concludere, allora, da decenni di neuroimaging?
Che il cervello ADHD mostra alterazioni misurabili, ma che queste sono sottili, eterogenee e fortemente modulate dall’età, dal sesso, dalle comorbilità e dal trattamento.
Che non esiste una “firma” neuroanatomica diagnostica, e che cercarla è probabilmente un obiettivo sbagliato — come cercare una firma radiologica della depressione o dell’ansia.
Che il valore dell’imaging risiede altrove: nel comprendere le traiettorie di sviluppo, nel predire esiti individuali, nel guidare scelte terapeutiche.
Il cervello, dopotutto, non è una mappa statica: è un sistema in costante movimento, che pensa, sente, agisce. E per capire l’ADHD dobbiamo guardare non solo a come è fatto, ma a come funziona — chimicamente, elettricamente, dinamicamente.
È nel territorio della neuropsicofarmacologia che i pezzi del puzzle iniziano a combinarsi in un quadro più coerente, dove la dopamina e la noradrenalina sono protagoniste.
Neuropsicofarmacologia: il ruolo centrale della dopamina e della noradrenalina
Se la neuroanatomia ci mostra dove il cervello ADHD differisce, la neuropsicofarmacologia ci suggerisce come — attraverso quali messaggeri chimici, quali circuiti, quali meccanismi molecolari.
Ed è qui che emerge un quadro sorprendentemente coerente: quasi tutti i farmaci efficaci per l’ADHD agiscono, direttamente o indirettamente, su due sistemi monoaminergici — la dopamina e la noradrenalina. Non è una coincidenza farmaceutica, ma una finestra sul disegno biologico del disturbo.
Stimolanti
Gli stimolanti — metilfenidato e anfetamine — sono i più antichi e i più potenti. Il loro meccanismo, studiato per decenni con tecniche da neurochimica molecolare, è elegante nella sua precisione.
Entrambi inibiscono i trasportatori di dopamina e noradrenalina (DAT e NET), le proteine di membrana che normalmente “riciclano” questi neurotrasmettitori dal sinapsi verso l’interno della cellula. Bloccando questo reuptake, stimolanti e anfetamine aumentano la concentrazione extracellulare di dopamina e noradrenalina, prolungando e amplificando la segnalazione sinaptica.
- Ma l’anfetamina va oltre: inibisce anche il trasportatore vescicolare VMAT2, spingendo il neurone a rilasciare più dopamina dalle vescicole di stoccaggio, e blocca l’attività della monoamino ossidasi (MAO), l’enzima che degrada le monoamine nel citoplasma. È un “attacco su più fronti” che spiega perché le anfetamine siano generalmente più potenti del metilfenidato, e anche perché il loro profilo di effetti collaterali sia più marcato.
- Il metilfenidato, più selettivo, agisce principalmente sull’inibizione dei trasportatori, con un’attività agonistica aggiuntiva sui recettori 5-HT1A serotoninergici e un’interazione con recettori alfa-2 adrenergici che modulano l’eccitabilità corticale.
| Farmaco | Meccanismo principale | Meccanismi secondari | Sito d’azione prevalente |
|---|---|---|---|
| Metilfenidato | Inibizione DAT e NET | Agonismo 5-HT1A; interazione alfa-2 adrenergica | Striato, ma anche corteccia e mesencefalo |
| Anfetamine (lisdexamfetamina, dexanfetamina) | Inibizione DAT e NET + rilascio vescicolare via VMAT2 + inibizione MAO | — | Striato (primario), corteccia, area tegmentale ventrale |
| Atomoxetina | Inibizione selettiva NET | Aumento indiretto dopamina nella corteccia prefrontale (NET presente anche su terminazioni dopaminergiche corticali) | Corteccia prefrontale (specifico) |
| Clonidina | Agonista alfa-2 adrenergico postsinaptico | Modulazione del locus coeruleus | Circuits noradrenergici diffusi |
| Guanfacina | Agonista alfa-2 adrenergico postsinaptico | Maggiore selettività per alfa-2A rispetto alla clonidina | Corteccia prefrontale (maggiore affinità) |
| Viloxazina | Inibizione reuptake noradrenalina | Modulazione serotoninergica (ruolo in ADHD ancora in esplorazione) | Sistema noradrenergico + serotonergico |
Non Stimolanti
La atomoxetina, primo non-stimolante approvato per l’ADHD e purtroppo non più in commercio in Italia, racconta una storia molecolare diversa ma complementare.
È un inibitore selettivo del trasportatore della noradrenalina (NET), senza azione diretta sul DAT. Eppure aumenta anche la dopamina nella corteccia prefrontale — come? Il segreto risiede nella distribuzione dei trasportatori: nella corteccia prefrontale, dove le terminazioni dopaminergiche esprimono NET oltre a DAT, bloccare il reuptake di noradrenalina influenza indirettamente anche la dopamina. È un esempio di come l’anatomia funzionale moduli la farmacologia: lo stesso meccanismo molecolare può avere effetti diversi a seconda del territorio cerebrale.
Questa selettività prefrontale spiega perché l’atomoxetina sia efficace su sintomi di inattenzione e disfunzione esecutiva, ma abbia effetti più modesti sull’iperattività motoria rispetto agli stimolanti — e perché non presenti lo stesso potenziale di abuso.
Più enigmatici sono gli agonisti alfa-2 adrenergici, clonidina e guanfacina. Entrambi stimolano recettori alfa-2 postsinaptici, ma i meccanismi neurali attraverso cui migliorano i sintomi ADHD restano, paradossalmente, non del tutto chiariti nonostante decenni di uso clinico.
Si ipotizza un’azione sul locus coeruleus, il nucleo pontino che proietta noradrenalina in tutto il cervello, modulando l’arousal e l’attenzione selettiva.
- La guanfacina, più selettiva per il sottotipo alfa-2A, nota anche per le sue proprietà antinfiammatorie, sembra avere un profilo più “corticale” e meno sedativo della clonidina, ma la traduzione da meccanismo molecolare a effetto clinico passa attraverso circuiti ancora da mappare completamente.
La viloxazina, anch’essa non più presente in commercio in Italia, aggiunge un capitolo intrigante: oltre all’inibizione del reuptake di noradrenalina, modula l’attività serotoninergica. Il ruolo esatto della serotonina nell’ADHD è ancora in fase di esplorazione — non è un neurotrasmettitore “core” come dopamina e noradrenalina, ma interagisce con entrambi nei circuiti di regolazione emotiva e impulsiva.
La viloxazina rappresenta un promemoria che la farmacologia dell’ADHD è ancora in evoluzione, e che molecole con profili “ibridi” potrebbero offrire vantaggi in sottogruppi specifici, ad esempio con comorbilità ansiose o disregolazione emotiva marcata.

| Farmaco | Bersaglio molecolare primario | Effetto su dopamina | Effetto su noradrenalina | Effetto su serotonina | Profilo clinico tipico |
|---|---|---|---|---|---|
| Stimolanti (metilfenidato, anfetamine) | DAT + NET | ↑↑↑ Striato e corteccia | ↑↑ Corteccia e sistema diffuso | Minimo (metilfenidato: agonismo 5-HT1A) | Efficace su tutti i sintomi core; rapido onset; potenziale di abuso |
| Atomoxetina | NET selettivo | ↑ Corteccia prefrontale (indiretto) | ↑↑ Sistema noradrenergico | Minimo | Efficace su inattenzione; lento onset (settimane); nessun potenziale di abuso |
| Alfa-2 agonisti | Recettori alfa-2 postsinaptici | Indiretto, non chiaro | Modulazione locus coeruleus | Minimo | Efficace su iperattività e impulsività; sedazione possibile; uso spesso in combinazione |
| Viloxazina | NET + modulazione 5-HT | Indiretto, modesto | ↑↑ | Modulazione | Profilo in evoluzione; possibile vantaggio in comorbilità emotive |
L’implicazione più profonda di questo panorama farmacologico è che l’efficacia dei farmaci che agiscono su dopamina e noradrenalina costituisce, di per sé, un argomento a favore dell’ipotesi di disregolazione di questi sistemi nel circuito fronto-striato-limbico.
È un ragionamento per “convergenza farmacologica”: se farmaci con meccanismi diversi ma convergenti su questi due neurotrasmettitori sono efficaci, è probabile che il disturbo coinvolga una disregolazione funzionale — non necessariamente un “deficit” quantitativo, ma un’alterazione della dinamica temporale e spaziale della segnalazione — in questi circuiti.
Il circuito fronto-striato-limbico, che connette corteccia prefrontale, nuclei della base, talamo e strutture limbiche come l’amigdala, è il teatro neurale dove dopamina e noradrenalina modulano motivazione, rinforzo, attenzione selettiva, inibizione comportamentale e regolazione emotiva.
Disregolare questo circuito significa, in termini clinici, perdere il filo delle priorità, cedere all’impulso del momento, faticare a procrastinare gratificazioni, sentirsi sopraffatto da stimoli emotivamente salienti.
| Evidenza a favore della disregolazione dopaminergica/noradrenergica | Limiti e cautele |
|---|---|
| Efficacia di farmaci che aumentano dopamina/noradrenalina | Gli stimolanti migliorano performance anche in soggetti sani (effetto “cognitivo” non specifico) |
| Correlazione genetica con vie dopaminergico e noradrenergico | I geni identificati spiegano solo una frazione della varianza; pathway pleiotropici |
| Alterazioni funzionali in fMRI durante compiti di inibizione | Differenze di attivazione non specifiche per ADHD (sovrapposizione con altri disturbi) |
| Effetti neuropsicologici specifici (memoria di lavoro, attenzione) | Miglioramenti farmacologici non sempre correlati a cambiamenti di qualità di vita |
| Modelli animali di ADHD (collico lesioni, DAT knockout) | Validità ecologica limitata; nessun modello cattura l’intero fenotipo umano |
C’è infine una domanda che la neuropsicofarmacologia solleva ma non risolve: perché, se la disregolazione dopaminergica è centrale, l’ADHD non risponde alla L-DOPA o ad altri potenziatori dopaminergici usati nel Parkinson?
E perché farmaci che aumentano la serotonina — gli SSRI — non sono efficaci sui sintomi core, pur essendo utili in comorbilità depressive?
Le risposte passano probabilmente attraverso la complessità dei circuiti: non è la quantità assoluta di dopamina che conta, ma la sua dinamica spazio-temporale, il rapporto tra fasi toniche e fasiche, la modulazione contestuale da parte della noradrenalina, l’interazione con altri sistemi. La farmacologia dell’ADHD, in altre parole, ci dice che il cervello non è un serbatoio di neurotrasmettitori da riempire o svuotare, ma un orchestra dove il tempo, l’armonia e il contrappunto contano tanto quanto il volume.
Bibliografia
Cortese S, Bellgrove MA, Brikell I, et al. Attention-deficit/hyperactivity disorder (ADHD) in adults: evidence base, uncertainties and controversies. World Psychiatry. 2025;24(3):347-371.
Sonuga-Barke, E. J., & Castellanos, F. X. (2007). Spontaneous attentional fluctuations in impaired states and pathological conditions: a neurobiological hypothesis. Neuroscience and biobehavioral reviews, 31(7), 977–986. https://doi.org/10.1016/j.neubiorev.2007.02.005